Imaging Pearls ❯ Stomach ❯ Gastric Polyps
|
-- OR -- |
|
- Gastric Polyps: Differential Dx
- Fundic gland polyps (FGP) are the most common polyp type comprising up to 77 % of all gastric polyps
- Hyperplastic polyps are the second most common type of gastric polyps in Western populations, comprise almost 15 % of all gastric polyps
- Gastric adenomas are the third most common type of polyp in the Western population, but only account for <1 % of all gastric polyps - Gastric Polyps: Syndromes
- familial adenomatous polyposis
- MUTYH-associated polyposis
- Lynch syndrome
- Peutz–Jeghers syndrome
- Juvenile polyposis syndrome
- Cowden (PTEN hamartoma tumor syndromes)
- Cronkhite–Canada syndrome 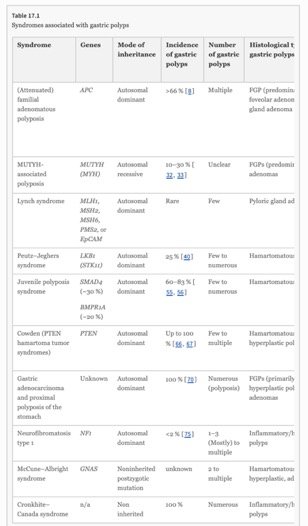
- Familial Adenomatous Polyposis Syndrome
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome caused by germline mutation in the Adenomatous Polyposis Coli (APC) gene. Classic FAP is characterized by development in teenage years of hundreds to thousands adenomatous polyps (≥100) throughout the colorectum. About 50 % of patients develop adenomas by age 15 and 95 % by age 35. If left untreated, CRC is diagnosed at an average age of 39 years (range 35–43 year). Attenuated FAP (AFAP) is defined by the presence of oligopolyposis; on average patients have 30 polyps. The diagnosis should be considered in patients 40–50 year old with 10–99 adenomas cumulatively. Patients with AFAP have a 70 % lifetime risk of CRC, presenting about 12 years later than classic FAP. - “More than two-thirds of FAP patients have gastric polyps. The vast majority (nearly 80 %) of gastric polyps in FAP are benign fundic gland polyps occurring in the gastric fundus and body. Typically, FAP patients have multiple FGPs presenting as gastric polyposis.”
Syndromic Gastric Polyps: At the Crossroads of Genetic and Environmental Cancer Predisposition.
Brosens L.A.A., Giardiello F.M., Offerhaus G.J., Montgomery E.A. (2016) In: Jansen M., Wright N. (eds) Stem Cells, Pre-neoplasia, and Early Cancer of the Upper Gastrointestinal Tract. "Advances in Experimental Medicine and Biology, vol 908. Springer, Cham. https://doi.org/10.1007/978-3-319-41388-4_17 - Peutz–Jeghers Syndrome
Peutz–Jeghers syndrome (PJS) is an autosomal dominant syndrome caused by germline mutation of the LKB1/STK11 gene. PJS is characterized by gastrointestinal polyposis, perioral pigmentation, and a moderate or high risk of a diversity of malignancies with an overall lifetime risk of any cancer of 81 % by age 70. Patients are particularly at increased risk for gastrointestinal malignancies, including colorectal, gastric, small bowel, and pancreatic cancer. In addition, increased risk for a variety of extraintestinal malignancies exists, including lung, breast, and gynecological cancer. - Peutz–Jeghers Syndrome
Polyps most frequently occur in the small intestine in about 95 % of PJS patients. The colon and stomach each are affected in about 25 % of patients. Guidelines recommend that upper and lower gastrointestinal endoscopies are performed first at age 8 years and repeated at least every 3 years when polyps are found. Small bowel imaging should be done at least every 3 years starting at age 8 years. Clearing of all polyps is recommended when possible. - Juvenile Polyposis Syndrome
Patients with JPS have an increased risk of several gastrointestinal malignancies. The lifetime risk of colorectal cancer has been calculated to be 38 % but may be as high as 70 %. In addition, JPS patients appear to be at increased risk of stomach, duodenal, and pancreatic cancer, but no formal risk analysis for these malignancies exists. Evaluation of literature reports suggests that gastric and small bowel carcinoma, together, occur at about one-fifth the frequency of colorectal cancers in this patient group. SMAD4 mutations are associated with a more aggressive gastrointestinal phenotype, involving higher incidence of colonic adenomas and carcinomas and more frequent upper gastrointestinal polyps and gastric cancer. - Cronkhite–Canada Syndrome
Cronkhite–Canada syndrome (CCS) is a rare protein-losing enteropathy typically characterized by diffuse gastrointestinal polyposis and typical ectodermal changes, such as hair loss and nail dystrophy. Although recent studies favor an autoimmune etiology, the precise cause of CCS has not been elucidated. More than 80 % of patients are diagnosed at age 50 or older. The prognosis is poor. Less than 5 % of patients have complete remission and a 5-year mortality rate of 55 % is noted due to gastrointestinal bleeding, sepsis, and congestive heart failure. There is no standard therapy but limited success has been reported with antibiotics, steroids, and partial gastrectomy. - Cronkhite–Canada Syndrome
Typically polyposis in CCS is diffuse throughout the entire gastrointestinal tract. The esophagus is uninvolved. The polyps are broad based and sessile and are a few millimeters to 1.5 cm in size. In the upper gastrointestinal tract, diffuse mucosal thickening rather than polyposis can be the main endoscopic picture, which may be more suggestive for gastric malignancy (lymphoma of linitis plastica) or gastric infection than CCS polyposis.